Perioperative Management of Carotid Body Paragangliomas
Sumeeta Kapoor, MD, Gifty Addae, MD, Maria Borrelli, DO, Robina Matyal, MD
Published April 18, 2025 | Clinics in Medical Education
Issue 6 | Volume 1 | April 2025
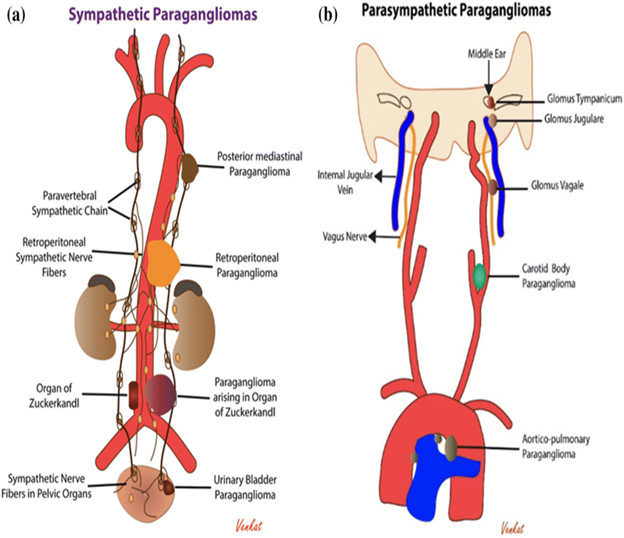
Paragangliomas: Overview and Classification: Paragangliomas are rare neuroendocrine tumors arising from extra-adrenal autonomic paraganglia, composed primarily of neuroendocrine cells derived from the embryonic neural crest. At the cellular level, paragangliomas and pheochromocytomas are indistinguishable. Catecholamine-secreting paragangliomas present similarly to pheochromocytomas, with symptoms such as hypertension, episodic headaches, sweating, and tachycardia.
Approximately 80%–85% of pheochromocytoma-paraganglioma (PPGL) cases are pheochromocytomas, while 15%–20% are paragangliomas. The overall prevalence of PPGL is approximately 6 cases per 1 million person-years. At least onethird of PPGL cases are associated with germline mutations and may be part of hereditary syndromes such as multiple endocrine neoplasia (MEN) type 2, von Hippel-Lindau (VHL) syndrome, and neurofibromatosis type 1 (NF1). As a result, genetic testing is recommended for all patients with PPGL.
The Shamblin classification, first introduced in 1970, is widely used for risk stratification in carotid body tumors (CBTs) before surgery. It categorizes tumors into three groups based on operative risk:
Type I: Small tumors that do not compromise the carotid vessels, allowing for relatively easy excision.
Type II: Tumors that adhere to or partially encase vessels, making excision more challenging.
Type III: Large tumors that intimately surround or encase the carotid vessels, posing significant surgical risk.
CBTs can be classified into three distinct forms: familial, hyperplastic, and sporadic. Familial cases are often associated with germline mutations in three of the four succinate dehydrogenase (SDH) subunit genes.
Case 1
36-year-old male presented with dysphagia, dysphonia and left upper eyelid ptosis for treatment of left carotid body tumor.
PMH: Hereditary paraganglioma pheochromocytoma syndrome, iatrogenic Horner’s syndrome and sleep apnea
Family history: Lung cancer and SDH-B mutation
MRI neck showed heterogeneous, enhancing mass at the left carotid bifurcation which splayed the internal and external carotid artery branches. This measures approximately 1.7 cm transverse by 1.7 cm AP by 4.3 cm
CTA head and neck: 2.2×1.7 cm cavernous malformation, left carotid space mass splaying the carotid vessels2.2×2.1×3.8cm
24h urine metanephrines and catecholamines were within normal limits.
Fiberoptic laryngoscopy: Clear nasal cavity bilaterally, clear nasopharynx, clear oropharynx, clear hypopharynx. Clear base of tongue, vallecula and bilateral piriform sinuses.
Preop: Successful embolization of the left carotid body tumor using particles and Gelfoam was performed, along with a negative balloon occlusion test of the left ICA, one day before surgery.
Intraop: General anesthesia with invasive arterial pressure monitoring, cerebral oximetry, and SedLine monitoring. The tumor appeared to originate from the sympathetic chain, requiring partial resection of the chain along with the left superficial laryngeal nerve. Lymph nodes were dissected and sent for biopsy to assess for lymphatic invasion.
Postop: Final histopathology report showed the lesion to be a malignant paraganglioma with perineural and lymphatic invasion.
Case 2
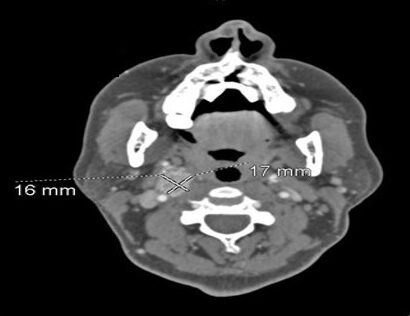
65-year-old female presented for evaluation of a right carotid body tumor. She was incidentally found to have a right carotid body tumor 2 years earlier during work-up for jaw pain.
PMH: HTN, HLD, carotid body tumor, sciatica, UTI, eczema
Urine metanephrines were normal.

Case 3
49-year-old Male with carotid body paraganglioma presented for resection of the tumor.
PMH: HTN, HLD, Bilateral carotid artery stenosis, family history of SDHB mutation
Intraoperatively patient had sustained BP of 280/180 for 8 min after induction despite large doses of antihypertensives and preop optimization. Patient had pulmonary edema after repeated spikes of hypertensive episodes.
CT neck: R carotid space mass with additional possible vertebral artery collateral, medialization of internal carotid artery to retropharyngeal location, 3.7 cm (2019: 3.0 cm)
Octreotide whole body scan in 2017: showed no uptake aside from bilateral carotid body tumors
- Hgb: 14.7
- Plt: 316
- Cr: 0.8
- Na: 139
- K: 4.4
- Cl: 100
- HCO3: 24
- AG:15
Normetanephrines: 1646 (88-640)
Metanephrines: 1720 (180-739)

Figure 1
Paraganglioma at the carotid bifurcation during neck dissection

Figure 2
Resected tumor

Figure 3
Carotid bifurcation after tumor resection
Postoperative Monitoring and Long-Term Follow-Up
Immediately after surgery, patients should be closely monitored for hemodynamic instability, including hypotension and arrhythmias, due to abrupt fluctuations in catecholamine levels. Postoperative hypotension is common and may require intravenous fluids and vasopressors for stabilization.
Biochemical monitoring is essential to assess the completeness of tumor resection. The North American Neuroendocrine Tumor Society recommends measuring 24-hour urine fractionated metanephrines and/or plasma-free metanephrines 2–8 weeks postoperatively. Persistent elevation may indicate residual tumor or metastatic disease.
Long-term follow-up is critical, particularly for patients with hereditary syndromes, due to the risk of recurrence or new tumor development. The National Comprehensive Cancer Network (NCCN) recommends regular biochemical testing and imaging, with follow-up intervals tailored to tumor characteristics and genetic risk factors. For sporadic, solitary tumors ≤5 cm, biennial follow-up is advised, whereas patients with larger, multiple, or hereditary tumors should undergo annual surveillance.

REFERENCES
1. Postoperative Management in Patients With Pheochromocytoma and Paraganglioma. Mamilla D, Araque KA, Brofferio A, et al. Cancers. 2019;11(7):E936. doi:10.3390/cancers11070936.
2. Perioperative Hemodynamic Instability in Pheochromocytoma and Sympathetic Paraganglioma Patients. Kim JH, Lee HC, Kim SJ, et al. Scientific Reports. 2021;11(1):18574. doi:10.1038/s41598-021-97964-3.
3. Araujo-Castro, M., Pascual-Corrales, E., Nattero Chavez, L. et al. Protocol for presurgical and anesthetic management of pheochromocytomas and sympathetic paragangliomas: a multidisciplinary approach. J Endocrinol Invest 44, 2545–2555 (2021).
4. Fagundes GFC, Almeida MQ. Perioperative Management of Pheochromocytomas and Sympathetic Paragangliomas. J Endocr Soc. 2022 Jan 14;6(2):bvac004.